)
Farbstoffe | Dietrich Zawischa Kontakt English version |
Was liegt der Farbigkeit zugrunde? Um der Antwort näher zu kommen, will ich zunächst die chemische Struktur von einigen Farbstoffen (im weiteren Sinn) besprechen.
|
| Indigofera heterantha Wallis, nahe verwandt und sehr ähnlich der Indigopflanze Indigofera tinctoria L., aus der der Farbstoff Indigo gewonnen wurde. Die grüne Farbe der Blätter wird – wie bei allen grünen Pflanzen – durch reichlich vorhandenes Chlorophyll hervorgerufen, die rosa Farbe der Blüten durch Farbstoff aus der Gruppe der Anthocyane, die später behandelt werden sollen. Der blaue Farbstoff Indigo ist in der Pflanze nur in einer Vorstufe, dem fast farblosen Indican, enthalten, das erst durch die beim Färbevorgang ablaufenden chemischen Prozesse zu Indigo umgewandelt wird. |
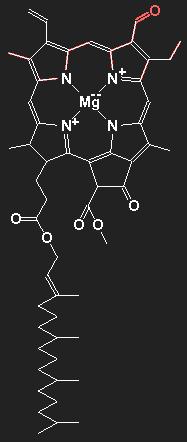 |
Chemische Strukturformel von Chlorophyll a (ohne das rot gezeichnete Teil) und Chlorophyll b (mit dem roten Anhängsel). Die Hauptbestandteile organischer Substanzen sind Kohlenstoff und Wasserstoff. Die Linien symbolisieren die chemischen kovalenten Bindungen, jede Linie repräsentiert also ein Paar von bindenden Elektronen. Doppellinien stehen für Doppelbindungen. Die Kohlenstoff-Atomrümpfe selbst werden der Übersichtlichkeit halber nicht eingezeichnet; an jedem Knick- oder Endpunkt der Linien befindet sich ein Kohlenstoffatom, wenn nicht ein anderes chemisches Symbol eingezeichnet ist. Außer Kohlenstoff und Wasserstoff sind am Aufbau des Moleküls noch Stickstoff N, Sauerstoff O und Magnesium Mg beteiligt. Die an die Kohlenstoffatomen gebundenen Wasserstoffatome werden (samt Bindungselektronen) ebenfalls nicht eingezeichnet. Kohlenstoff tritt immer vierwertig auf; gehen von einem (Kohlenstoff-) Punkt weniger als vier Linien aus, so muss man sich statt der fehlenden Linien angebundene Wasserstoffatome denken. Eine frei endende Linie steht also für eine CH3-Gruppe. Das Chlorophyll b unterscheidet sich vom Chlorophyll a dadurch, dass eine Methylgruppe / durch eine Formylgruppe /=O ersetzt ist. |
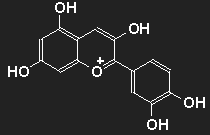 | Cyanidin als Beispiel für einen Farbstoff aus der Gruppe der Anthocyane. Durch Wegfall von hier vorhandenen oder Hinzukommen von weiteren OH- oder auch OCH3-Einheiten ergibt sich eine Fülle von verschiedenen Farbstoffen in den Bereichen Blau über Violett und Purpur bis Rot. |
) | Die Kornblume Centaurea cyanus L. verdankt ihre Farbe dem Cyanidin. |
Carotinoide sind Farbstoffe pflanzlichen Ursprungs mit einander ähnlichem Aufbau. Rote, orange und gelbe Farben von Blüten, Früchten und Wurzeln werden durch Carotinoide hervorgerufen; in grünen Pflanzen ist neben Chlorophyll auch Carotin enthalten.
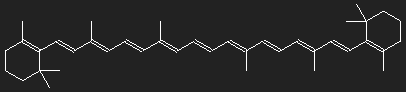
Die Strukturformeln geben nicht genau den räumlichen Aufbau wieder – die dreidimensionalen Strukturen werden in die Zeichenebene gelegt und dadurch verzerrt. Sie geben einen gewissen Einblick in die Bindungsverhältnisse. Aber auch in dieser Hinsicht ist die Wirklichkeit nur schwer zu Papier zu bringen.
Was auffällt, ist die große Zahl von Doppelbindungen zwischen den Kohlenstoffatomen. In den meisten Fällen wechseln sich auf längeren Wegstrecken entlang des Molekülgerüsts Einfach- und Doppelbindungen ab. Dies ist, wie wir sehen werden, für die Farbigkeit wichtig.
 |
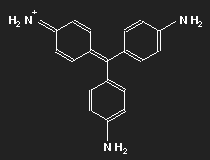 |
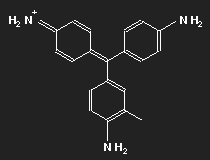
| Fuchsin | Parafuchsin |
Fuchsin ist nach dem Mauvein der zweite synthetisch hergestellte Farbstoff. Benannt wurde er nach einer aus Amerika stammenden Zierpflanze, der Fuchsie; andere Namen sind Magenta und Rosanilin. Meist ein Gemisch aus einander ähnlichen Substanzen, Fuchsin und Parafuchsin, letzteres mit etwas einfacherer, symmetrischer Struktur.
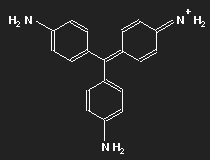
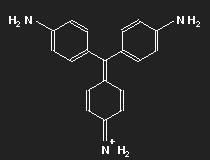
Betrachten wir das Parafuchsin. Der Farbträger ist ein positives Ion. Bei gegebener räumlicher Anordnung der Konstituenten und unter Berücksichtigung ihrer bekannten Wertigkeiten ergibt sich eine Strukturformel wie oben links gezeigt. Oder wie im mittleren Bild – oder wie oben rechts. Die mittlere Form ist genau das Spiegelbild der linken und lässt sich durch Drehung in diese überführen, ebenso die dritte. Die positive Ladung befindet sich am Stickstoffatom links oben – oder rechts oben – oder unten.
Tatsächlich entscheidet sich die Natur nicht zwischen diesen Möglichkeiten: der Zustand geringster Energie ist eine Überlagerung aus allen dreien. In diesem Zustand findet auch keine innermolekulare Bewegung der Elektronen statt (sonst müsste ja elektromagnetische Strahlung ausgesandt werden). Hier äußert sich wieder die Besonderheit der Quantenmechanik, die es uns schwierig – genauer gesagt: unmöglich – macht, sie gefühlsmäßig anschaulich in den gewohnten Begriffen zu verstehen. Ein unteilbares Teilchen kann sich auf verschiedene Orte verteilen.
Das heißt für die alternierenden Doppelbindungen, dass auch sie nicht fest lokalisiert, sondern entlang der Kette etwas diffuser verteilt sind, "delokalisiert" nennt man das. Das ist im Fall des Benzolringes wohlbekannt: alle Bindungen zwischen den sechs C-Atomen sind gleichartig, das wird oft durch ein spezielles Zeichen (ein einfaches Sechseck mit einem darinliegenden Kreis) deutlicher gemacht.
Zur Vereinfachung der Diskussion betrachten wir zunächst ein fiktives System, in dem für das Elektron nur zwei Aufenthaltsorte möglich sind: dessen quantenmechanischer Grundzustand lässt sich dann schreiben als
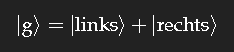
Die Überlagerung
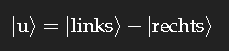
gibt es auch, dieser Zustand hat eine etwas höhere Energie. Als Zustand fester Energie Eu hat er laut Quantenmechanik die Zeitabhängigkeit

(h ist das Plancksche Wirkungsquantum). Diese "triviale" Zeitabhängigkeit hat für den Zustand fester Energie Zeitunabhängigkeit aller messbaren Größen zur Folge. Auch hier bewegt sich nichts.
Bewegung tritt erst auf, wenn Zustände verschiedener Energie überlagert werden, als Beispiel die Zustände |g⟩ und |u⟩ mit gleichen Gewichten. Die Rechnung ist einfach, und das Ergebnis lässt sich in eine Form bringen, der man ansieht, dass eine Oszillation zwischen |links⟩ und |rechts⟩ stattfindet, mit anderen Worten: die Ladung fließt zwischen den beiden Aufenthaltsorten hin und her, entlang der aus Kohlenstoffatomen mit den alternierenden Einfach- und Doppelbindungen gebildete Wegstrecke kann Strom fließen.
Der Übergang vom Zustand |g⟩ in den Zustand |u⟩ kann unter Absorption, der umgekehrte unter Emission eines Lichtquants erfolgen. Man kann sich vorstellen, dass während des Absorptions- bzw. Emissionsvorganges eine Überlagerung von Anfangs- und Endzustand vorliegt, bei der sich die Ladung genau mit der passenden Frequenz hin und her bewegt. Dieser Ladungstransfer über mehrere Atomabstände hat große Emissions- und Absorptionswahrscheinlichkeiten zur Folge.
Im Fall von drei verschiedenen Aufenthaltsorten wie beim Parafuchsin (und Fuchsin) wird es etwas komplizierter: mit einfachen Modellannahmen ergeben sich nach kurzer Rechnung die (nicht normierten) Zustände (in naheliegender Notation)
Ketten mit abwechselnden Einfach- und Doppelbindungen finden sich in verschiedenen Formen in den Farbstoffmolekülen. Im einfachsten Fall sind sie mehr oder weniger gerade, nur etwas zackig. Zum Beispiel im Carotin. Lassen wir zur Vereinfachung die Ringstrukturen an den Enden und die an der Kette sitzenden CH3-Gruppen weg, vielleicht haben sie ja nur die Funktion von Griffen und Henkeln zur Manipulation durch die molekularen Maschinen und beeinflussen das Absorptionsvermögen nur wenig. Um zu einer groben Abschätzung des Absorptionsvermögens zu kommen, ignorieren wir alle Details der Struktur.
Wir setzen voraus, dass es die zu den Doppelbindungen führenden Elektronen sind, auf die es ankommt, und denken uns diese Elektronen zunächst alle entfernt. Zurück bleibt eine Kette von 22 Kohlenstoffatomen, die mit Einfachbindungen aneinander gebunden sind. An den Enden sitzen jeweils zwei H-Atome, alle anderen C-Atome haben je ein H-Atom gebunden. Pro Doppelbindung haben wir zwei Elektronen entfernt, somit befindet sich an jedem Kohlenstoffatom eine positive Überschussladung. Das ist das "passive" Grundgerüst des Farbstoffmoleküls, das wir uns einfach durch einen langgestreckten Potentialtopf oder Kasten ersetzt denken. In diesen Kasten sind die fehlenden Elektronen wieder einzufüllen. Wir vernachlässigen die Wechselwirkung der Elektronen untereinander.
Die Form der Wellenfunktionen wäre nun mit Hilfe der Schrödingergleichung zu bestimmen. Wir können annehmen, dass die Wellenfunktionen in den Richtungen senkrecht zur Längsachse des Moleküls alle gleich sind, dann brauchen wir uns um diese Richtungen nicht mehr zu kümmern und haben die Aufgabe schließlich auf ein eindimensionales Problem reduziert: ein eindimensionaler Potentialtopf mit undurchdringlichen Wänden, innerhalb dessen sich die Elektronen frei bewegen können.
In diesem Fall kennen wir die Lösungen der Schrödingergleichung schon: die Wellenfunktionen haben die gleiche Form wie die Eigenschwingungen einer an beiden Enden eingespannten Saite. Da sich die Elektronen innerhalb des Kastens frei bewegen können, können wir die Beziehungen zwischen Wellenlänge, Impuls und kinetische Energie für freie Teilchen verwenden: p = h/λ, E = p2/(2m). (p steht für den Impuls, λ für die Wellenlänge, h ist das Plancksche Wirkungsquantum, E die Energie und m die Masse der Elektronen.)
Bezeichnen wir die Länge des Kastens mit L, dann sind die möglichen Längen der stehenden Wellen der Reihe nach 2L, L, 2L/3, L/2, usw., also 2L/n, wobei n = 1, 2, 3 ... Die Quantenzahl n ist einfach die Nummer des Einteilchenzustandes. Die zugehörigen Energien sind En = h2/(8 m L2) n2.

Jeder der eben berechneten Einteilchenzustände kann wegen des Pauliprinzips nur zwei Elektronen mit entgegengesetzt gerichteten Spins aufnehmen. Wenn 22 Elektronen untergebracht werden, dann ist im Zustand niedrigster Energie das Niveau mit der Nummer 11 zweifach besetzt, das mit der Nummer 12 leer. Durch die Einwirkung elektromagnetischer Strahlung kann ein Elektron aus Niveau Nr. 11 in Niveau 12 "angehoben" werden, dazu die Energie ΔE = E12 – E11 nötig – mit anderen Worten: Lichtquanten, die diese Energie mitbringen, können absorbiert werden.
Mit Hilfe der Beziehung E = h ν = h c/λ, die für Lichtquanten gilt, können wir daraus die Wellenlänge λ der elektromagnetischen Strahlung am Maximum der Absorption gewinnen.
Wählen wir als Länge des Potentialtopfes die Zahl der Kohlenstoffatome, multipliziert mit dem typischen Abstand zweier C-Atome, der in Ketten mit alternierenden Doppelbindungen ca. 0.139 nm beträgt, so entspricht dies einer gestreckten Kette und berücksichtigt noch nicht den Sachverhalt, dass die chemischen Bindungen beim Kohlenstoff einen Winkel von etwa 120º einschließen. Wir wählen daher die effektive Länge noch um einen Faktor 0.866 kleiner (0.866 ist die Höhe in einem gleichseitigen Dreieck mit Seitenlänge 1).
Die so für einen Elektronen-Übergang von Niveau 11 nach 12 benötigte Photonenenergie entspricht einer Wellenlänge der elektromagnetischen Strahlung von 1006 nm, das ist außerhalb des sichtbaren Bereiches im Infraroten. Trotz aller Näherungen – es ist zu erwarten, dass Carotin im Infraroten ein Absorptionsmaximum hat.
Kann Carotin auch Photonen mit höherer Energie absorbieren? Dies müsste dann zu Elektronenübergängen nicht zwischen benachbarten, sondern zwischen weiter auseinanderliegenden Energieniveaus führen. Der Übergang von Niveau 10 nach 12 oder von 11 nach 13 ist aus Symmetriegründen nicht möglich. Für die elektronische Anregung von Niveau 9 nach 12 berechnen wir λPhoton = 367 nm, also eine Wellenlänge knapp außerhalb des Sichtbaren im Ultraviolett. Offensichtlich ist unser Modell zu grob und die weggelassenen "Anhängsel" haben doch mehr Einfluss auf die Lage der Energieniveaus.
Das Modell reproduziert die Annahme, von der wir ausgegangen sind, dass nämlich die Elektronen, welche die Doppelbindungen ausmachen, über die ganze Kette alternierender Bindungen frei beweglich sind: die Dichte jedes einzelnen ergibt sich als über die ganze Kette verteilte Funktion. Die Kette mit alternierenden Doppelbindungen verhält sich somit analog wie ein elektrischer Leiter, wie ein winziges Drähtchen.
Man kann in einem Stück Draht (Antenne) durch Einstrahlen elektromagnetischer Wellen die Ladungsträger zu periodischen Schwingungen anregen. Die dazu nötige Frequenz ist umso höher, je kürzer der Draht ist. Lange Saiten geben tiefere Töne als kurze, Orgelpfeifen entsprechend, der Zusammenhang zwischen Länge und Schwingungsfrequenz bei sonst gleichen Bedingungen gilt ganz allgemein. Das von uns betrachtete quantenmechanische Modell verhält sich genauso. Die niedrigste (und am leichtesten anregbare) Schwingungsfrequenz ergab sich unterhalb der Frequenzen des sichtbaren Lichtes; Frequenzen im sichtbaren Bereich sind bei etwas kürzeren Ketten zu erwarten.
Versuchen wir die Abschätzung noch einmal bei einer etwas kürzeren Kette.
Carotin (Provitamin A) kann vom Körper in Vitamin A umgewandelt werden, aus diesem wird das Retinal, der farbaktive Teil des Sehpurpurs, synthetisiert. Der Sehpurpur besteht aus einem großen Eiweißmolekül Opsin, an das das Retinal gebunden ist. An der Strukturformel erkennt man noch deutlich die "Verwandtschaft" mit dem Carotin.
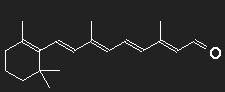
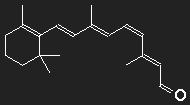
β-Carotin in der Mitte durchgeschnitten, der fehlende Teil durch ein Sauerstoffatom ersetzt – fertig ist das Retinal (zumindest auf dem Papier).
Feine Details können wir mit unserer groben Abschätzung ohnehin nicht berücksichtigen; dass am Ende der Kette kein Kohlenstoff, sondern hier ein Sauerstoff sitzt, wird, wie auch der Knick in der Kette beim 11-cis-Retinal, ignoriert. Betrachten wir einen Potentialkasten mit einer Länge, die einer Kette von 12 C-Atomen entspricht, in den 12 Elektronen einzufüllen sind. Bei der Anregung niedrigster Energie muss ein Elektron vom sechsten in das siebente Einteilchenniveau befördert werden, die dazu nötige Energie entspricht einer Wellenlänge λPhoton = 529 nm.
Das Absoptionsmaximum des Sehpurpurs liegt bei ca. 500 nm! – Dass wir mit unserer Abschätzung so nahe an diese Zahl gekommen sind, ist eher als Zufall zu werten. Aber dass wir die richtige Größenordnung erhalten haben ist wichtig.
Exakte Berechnungen der Eigenschaften von Molekülen sind nicht möglich, man ist immer auf numerische Näherungsverfahren angewiesen. Allen Verfahren gemeinsam ist, dass Wellenfunktionen von Elektronen in einem fiktiven (oder effektiven) Potential ermittelt werden, zu dem auch die Wechselwirkung der Elektronen untereinander beiträgt. Das Potential hängt von der räumlichen Verteilung der Elektronenwolken ab, also vom gesuchten Endergebnis. Man startet mit einer vermuteten Dichteverteilung, berechnet daraus das Potential, daraus wieder die Dichteverteilung, mit Hilfe derer das Potential neu berechnet wird. Das Verfahren ist so lange zu wiederholen, bis das Endergebnis mit genügender Genauigkeit die Eingabedaten reproduziert. (Hartree-Fock-Verfahren und Energiedichte-Funktionalmethode in verschiedenen Varianten.)
Eine weitere Komplikation ist, dass es nicht ausreicht, ein isoliertes Molekül zu berechnen, sondern dass man auch die Umgebung mit berücksichtigen muss. Die elektrischen Kräfte sind langreichweitig, daher spielt auch eine Rolle, was sich in der Nachbarschaft befindet. Es ist nicht einfach. Solche Rechnungen sind überhaupt erst durch leistungsfähige Computer/Großrechner möglich geworden.
Zum Glück ist die Chemie nicht auf solche Berechnungen angewiesen gewesen; die ersten künstlichen Farbstoffe wurden ja lange vor der Entdeckung der Quantenmechanik synthetisiert. Aber heute hilft uns die Quantenmechanik, zu verstehen, was den empirischen Regeln der Farbstoffchemie zugrunde liegt.
Um abzuschätzen, wie sich z.B. elektrische Ladungen (Ionen) in der Nähe des Farbstoffmoleküls auf dessen Farbe auswirken, betrachten wir noch einmal das Kastenpotentialmodell, und als Beispiel speziell den Übergang vom Niveau mit der Nummer 6 in das nächsthöhere mit der Nummer 7. Im Bild unten sind noch einmal die zugehörigen Dichteverteilungen gezeigt. Fügen wir jetzt in der Mitte des Kastens eine flache Potentialschwelle ein (die Schwelle soll niedrig im Vergleich mit der Energiedifferenz der betrachteten Elektronenzustände sein), dann wird das Elektron im unteren Zustand von der Schwelle kaum etwas fühlen, da es sich in diesem Bereich kaum aufhält, im oberen Zustand sitzt der Mittelpunkt eines der Wölkchen mitten auf der Schwelle, daher verschiebt sich die Energie dieses Zustandes stärker nach oben.

Dadurch vergrößert sich die Energiedifferenz zwischen den beiden Niveaus und das Maximum der Absorption verschiebt sich entsprechend zu kürzeren Lichtwellenlängen.
Wenn an einer Kette von C-Atomen mit alternierenden Doppelbindungen an einer Stelle ein H-Atom durch ein anderes Atom, eine OH-, CH3- oder eine andere Gruppe ersetzt wird, so hat dies eine lokale Veränderung des Potentials zur Folge, die sich ähnlich auswirken kann.
| |
 |
|
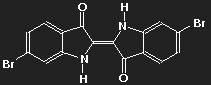
Indigo (Farbe des Pflanzenextrakts auf Papier oder Baumwolle) | Purpur (6,6'-dibromoindigo) (Farbe von Tyrischem Purpur) |
Der farbgebende Teil beider Moleküle (das "Chromogen") ist derselbe, nämlich die Kette konjugierter Doppelbindungen, die zwischen den beiden Sauerstoffatomen über vier Kohlenstoffatome verläuft. Das Potential, in dem sich die delokalisierten Elektronen bewegen, hängt jedoch von der Verteilung aller Ladungen im Molekül ab, und die ändert sich, wenn Wasserstoff durch Brom ersetzt wird.
Atomare Spektren nennt man Linienspektren, weil sie im Spektroskop wie eine diskrete Folge von Linien erscheinen – Linien deshalb, weil man das Bild des Spaltes, durch den das Licht in das Spektroskop eintritt, abhängig von der Wellenlänge an verschiedenen Stellen als scharfe Linien wahrnimmt. Mit hochauflösenden Geräten stellt man fest, dass es doch nicht nur eine scharfe Frequenz bzw. Wellenlänge ist, die jeweils zu einer Linie führt, sondern dass die Linien eine zwar geringe, aber doch endliche Breite haben. Die theoretische untere Grenze der Linienbreite, genannt "natürliche Linienbreite", wird im sichtbaren Bereich nie erreicht, die beobachtete, meist geringe Breite rührt vom Doppler-Effekt infolge der Wärmebewegung und von der sogenannten Stoßverbreiterung.
Als Stoß bezeichnet man den Vorgang, wo zwei Atome oder Moleküle sich nahe kommen, miteinander in Wechselwirkung treten und dadurch ihren Impuls (Bewegungsrichtung und Geschwindigkeit) ändern. Die Wechselwirkung mit dem Stoßpartner erfährt das Molekül wie ein kurzzeitig wirkendes Kraftfeld, das anziehend oder abstoßend sein kann, und das, wie oben besprochen, zu einer Verschiebung der Energieniveaus gegeneinander führt. Diese nur statistisch zu erfassenden Veränderungen der Übergangsfrequenzen äußern sich in einem nicht zu sehr verdünnten Gas als Stoßverbreiterung der Linie.
Die Spektren von Molekülen unterscheiden sich deutlich von den atomaren Linienspektren. Betrachten wir zunächst den einfachsten Fall eines zweiatomigen Moleküls. Das "hantelförmige" Molekül kann vibrieren und um seinen Schwerpunkt rotieren. Die typische Anregungsenergien von Schwingungen und Rotationen sind sehr viel kleiner als die Energien, die für elektronische Anregungen nötig sind. Elektronische Übergänge geschehen schnell im Vergleich zu den langsamen Schwingungen. Grob gesprochen bedeutet das, dass ein Schwingungszustand bestehen bleiben kann, während ein elektronischer Übergang erfolgt. Die Energien der elektronischen Niveaus hängen aber vom Abstand der Kerne ab; das führt letztlich dazu, dass für einen elektronischen Übergang nicht nur eine Energie, sondern eine diskrete Folge von etwas verschiedenen Energien (und somit Wellenlängen des Lichtes) beobachtet wird. Entsprechendes gilt für die Rotationszustände, die zu einer weiteren, noch feineren Struktur des Spektrums führen: statt einzelner Linien treten Vibrations- und Rotationsbanden auf.
Mehratomige Moleküle haben ein reichhaltigeres Schwingungsspektrum, das bald unübersichtlich wird. Je größer die Moleküle, desto dichter liegen auch die Rotationszustände, und das führt aufgrund der endlichen Linienbreite dazu, dass sich die einzelnen Linien in den Banden überlappen und nicht mehr getrennt werden können, insbesondere auch, weil die Moleküle in einem Lösungsmittel oder einer organischen Struktur mit ihren Nachbarn in Wechselwirkung stehen und aufgrund der thermischen Bewegung ununterbrochen Energie untereinander austauschen. Statt einzelner Emissions- oder Absorptionslinien wie bei Atomen beobachtet man daher bei den großen Molekülen sehr breite Absorptionskurven. Emissionsspektren von Farbstoffen sind nur in Ausnahmefällen zu beobachten, nämlich bei fluoreszierenden Farbstoffen.
Der Erscheinung der Fluoreszenz und verwandten Phänomenen ist ein eigener Abschnitt "Lumineszenz" gewidmet.
) |
Die Kermesbeere enthält einen purpurroten Farbstoff in den Beeren in so hoher Konzentration, dass diese schwarz sind. Der Farbstoff kann Licht im ganzen sichtbaren Bereich absorbieren; das Maximum der Absorption liegt bei mittleren Wellenlängen (grün). Im Stengel ist die Konzentration sehr gering, aber dies ergibt eine kräftige Farbe, weil der Farbstoff sehr stark absorbiert. Warum das so ist, soll im folgenden besprochen werden.
Asiatische Kermesbeere (Phytolacca acinosa Roxburgh) |
Eine beschleunigte elektrische Ladung sendet elektromagnetische Strahlung aus. Verhältnismäßig leicht zu behandeln ist der Fall, dass sich eine Ladung periodisch mit konstanter Frequenz hin und her bewegt, wobei die Amplitude der Oszillation sehr klein gegenüber der Wellenlänge der ausgesandten Strahlung ist. Entsprechend kann eine durch "elastische" Kräfte an ihre Ruhelage gebundene elektrische Ladung durch elektromagnetische Wellen zum Mitschwingen gebracht werden – die Energie der Schwingung wird der Strahlung entzogen, es wird also Strahlung absorbiert.
Die wesentliche Größe, von der das Ausmaß der Abstrahlung abhängt, ist das Dipolmoment, das ist das Produkt aus Ladung mal Auslenkung bzw. Schwingungsamplitude. Im Fall der Absorption ist die wesentliche Größe die Polarisierbarkeit, also die Größe des Dipolmoments, das von einer Einheit der elektrischen Feldstärke hervorgerufen wird.
In den quantenmechanischen Ausdrücken für die Emissions- und Absorptionswahrscheinlichkeit von elektromagnetischer Strahlung tritt das Dipol-Übergangsmatrixelement auf, das ist die Größe, die dem klassischen Dipolmoment entspricht. Wichtig in diesem Zusammenhang ist, dass die Formeln für Absorption und Emission das Dipolmatrixelement quadratisch enthalten. Ein zehnmal größeres Dipolmoment hat hundertmal soviel Absorption zur Folge. Und außerdem ist für unsere Überlegungen wichtig, dass wir die Größenordnung der Dipolmatrixelemente abschätzen können, wenn wir die Größe der betrachteten Moleküle kennen.
Für das einfache Modell des langgestreckten Kastenpotentials der Länge L lässt sich das Übergangs-Dipolmatrixelement Pm,n leicht berechnen, und diesen Wert können wir als groben Schätzwert für eine Kette konjugierter Doppelbindungen der gleichen Länge auffassen.
Zu berechnen ist für den Übergang eines Elektrons vom Einteilchenzustand n in den Zustand m
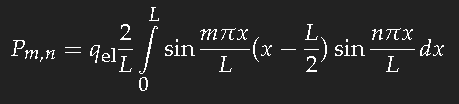
(qel ist die Ladung eines Elektrons.)
Die Rechnung ist nicht schwer. Falls m-n eine gerade Zahl ist, ist das Ergebnis Null; für m-n ungerade erhält man
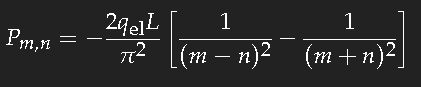
also für den Übergang zwischen zwei aufeinanderfolgenden Zuständen ungefähr 20 Prozent der Länge L multipliziert mit der Elektronenladung. (Das Vorzeichen ist unerheblich, denn in den Wahrscheinlichkeiten tritt immer nur das Quadrat dieser Größe auf.)
So grob sie ist: diese Abschätzung gibt uns auch noch für ein einzelnes Atom einen Richtwert, wenn wir für L den Durchmesser des Atoms annehmen, also ungefähr den Abstand zweier Atome in der chemischen Bindung.
Die mögliche Größe des Dipolmatrixelements ist also durch die Größe des Moleküls oder, genauer, durch die Ausdehnung der in dem Molekül vorliegenden Kette konjugierter Doppelbindungen beschränkt, beziehungsweise durch die atomaren Abmessungen. Infolge ihrer Größe können die Farbstoffmolekule elektromagnetische Strahlung – die Lichtquanten – sehr viel besser einfangen als einzelne Atome. So kommt ihre starke Färbekraft zustande.
Die entlang einer Kette konjugierter Doppelbindungen in einem Molekül delokalisierten Elektronen sind primär für das Absorptionsvermögen und somit für die Farbigkeit verantwortlich. Man nennt die entsprechenden Teile der Moleküle daher Chromophore, "Farbträger" oder Chromogene, "Farbquellen". Molekülbausteine, welche Elektronen leicht abgeben oder aufnehmen können (Elektronendonatoren und -Akzeptoren) haben große Dipolmomente zur Folge, wenn sie sich weit außen am Chromogen befinden und heißen daher Auxochrome, "Farbverstärker". Als Beispiel seien die NH2-Gruppen im Fuchsin genannt.
Bliebe noch etwas über die verschiedenen Arten von Textilfarbstoffen zu sagen, chemische Aspekte der Herstellung der Farben und den Färbevorgang, aber darüber gibt es ausreichend Literatur auch im Netz. Hier als Beispiel ein Link über Farbchemie.


Sie finden im Internet Angaben über die Herkunft und historische Entwicklung anorganischer Pigmente, Listen mit den gängigen Pigmenten, wie sie für Malerei und Herstellung von Farben verwendet werden, und Angaben über deren chemische Zusammensetzung. Zum Beispiel hier: Farbmittel und natürlich auch in der Wikipedia. Diesen Zusammenstellungen können Sie auch entnehmen, was sich hinter den Namen der im obigen Bild gezeigten Pigmente verbirgt, und Sie finden dort auch Angaben über die Ursachen der Farbigkeit. Ich will das nicht duplizieren und hier nur auf einen wichtigen Unterschied zu den Farbstoffen hinweisen:
An dem Bild oben ist es schon zu sehen: während bei den organischen Farbstoffen verschwindend kleine Substanzmengen schon für kräftige Farbe sorgen, ist bei den Pigmenten das Volumen und Gewicht durchaus nicht zu vernachlässigen, man sagt, die Pigmente "haben Körper".
Dies ist nicht nur für die praktische Anwendung wichtig, sondern auch für das theoretische Verständnis der Farbentstehung. Während man beim Farbstoff das Absorptionsvermögen eines einzelnen Moleküls betrachtet kann, hat man bei den Pigmenten die Farbigkeit eines zwar nur staubkorngroßen, aber auf der atomaren Skala riesigen Kristalls zu untersuchen. Dies erfordert andere Überlegungen und erfolgt daher in eimer getrennten Abhandlung über farbige Kristalle/Mineralien.
Zurück zur Übersicht oder zur Eingangsseite